
Promising treatment for Alexander disease moves from rat model to human clinical trials
Alexander disease is a progressive and rare neurological disorder with no cure or standard course of treatment. But a new study led by researchers at the University of Wisconsin–Madison involving a rat model of the disease offers a potential treatment for the typically fatal condition.
It’s a significant step in efforts to help people with the disease, says UW–Madison Waisman Center senior scientist Tracy Hagemann, who led the study alongside Albee Messing, professor emeritus of comparative biosciences and founder of the Alexander Disease Lab. With University of Alabama at Birmingham colleague Michael Brenner, Messing discovered the gene responsible for Alexander disease more than 20 years ago.
People born with Alexander disease may develop an enlarged brain and head, experience seizures or delayed development, have stiffness in their arms and legs, and have intellectual disabilities. The disease, which involves destruction of the white matter of the brain, is often not diagnosed until symptoms are pronounced, says Hagemann.

Tracy Hagemann

Albee Messing
The new study, published Nov. 17 in Science Translational Medicine, provided preliminary data instrumental for a human clinical trial currently being led by Ionis Pharmaceuticals. Hagemann, Messing, and the Alexander Disease Lab are not directly involved.
However, working with Ionis Pharmaceuticals, the researchers developed a treatment that consists of small pieces of DNA called antisense oligonucleotides, which in their rat model was able to target mRNA in cells and tag the mRNA for destruction, effectively halting it from creating proteins.
One feature of Alexander disease is the formation of abnormal protein aggregates called Rosenthal fibers, caused by mutations in the gene that makes a protein called GFAP. The connection between this abnormal GFAP and the white matter destruction seen in Alexander disease is not yet clear, but changes in the protein are an intrinsic part of the disease in almost all cases.
Studies with a mouse model developed by Hagemann, Messing and their collaborators, and published three years ago, showed that antisense oligonucleotides were able to reduce GFAP and clear Rosenthal fibers. However, mice display only subtle symptoms of Alexander disease and researchers can’t measure important improvements in behavior or quality of life that may result from treatment.
The research team was able to develop a rat model that better represents the white matter damage and physical manifestations seen in humans. The model also provides better opportunities to assess symptom improvement in response to antisense oligonucleotide treatment.
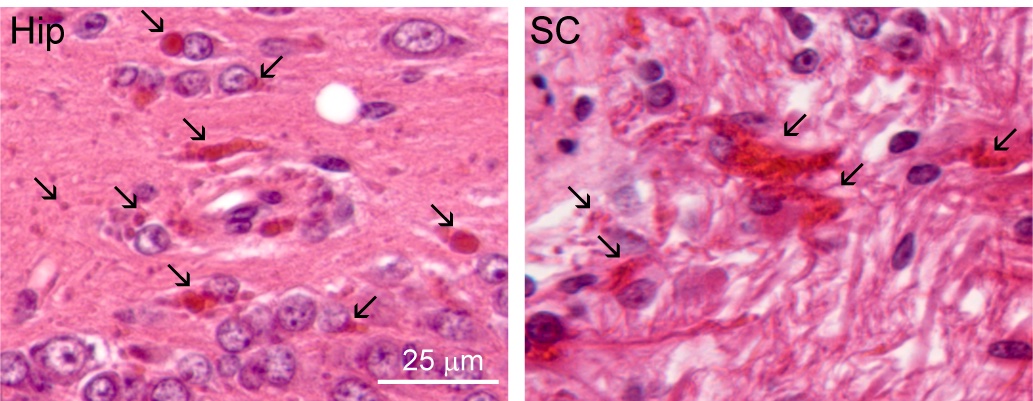
A hallmark of Alexander disease is the buildup of abnormal proteins called Rosenthal fibers, pictured here (red) in brain tissue from a study of a rat model of the disease. The study identified a potential treatment and helped provide early data for a human clinical trial now underway. Tracy Hagemann et al.
“Alexander disease is considered a leukodystrophy, where white matter deficits develop, and we don’t see evidence of that or motor impairment in the mouse model,” says Hagemann. “So, for a preclinical model, the rats are much improved compared to the mice.”
The rats treated with antisense oligonucleotides before they developed major physical symptoms stayed virtually indistinguishable from their healthy littermates. When treatment began after the rats were severely impaired, their symptoms not only drastically improved; they also experienced a reversal in some of the damage to their white matter.
The antisense oligonucleotides, she explains, “clear out the GFAP aggregates (or Rosenthal fibers), and not only can we prevent the disease from happening by treating animals at an early stage before they’re really showing significant clinical signs, we can treat them when they’re at their worst and see reversal of some of the disease phenotypes.”
In people, Hagemann says, “we’ll be happy if we can stop the disease from progressing. But if you can actually see some reversal of symptoms that have already occurred, that would be wonderful.”
In addition to creating a foundation for clinical trials, the rat model has also paved the way to study aspects of the disease that are not yet understood, Hagemann notes, including the first opportunity to study the link between the GFAP mutations and white matter deficits in mammals.
These developments have been possible because of Messing’s extensive work on Alexander disease over the last 25 years, as well as the contributions of colleagues around the world, Hagemann adds. Messing’s “dedication and commitment to Alexander disease research has deepened our understanding of the disorder immensely.”
Study co-authors include Robert F. Berman of the University of California, Davis; Mel B. Feany at Brigham and Women’s Hospital, Harvard Medical School and Boston Children’s Hospital; and Ming-Der Perng of National Tsing Hua University. The study was supported by grants from the National Institutes of Health, including the National Institutes of Health’s Eunice Kennedy Shriver National Institute of Child Health and Human Development (HD076892, HD03352, HD090256, and HD103526) and the National Institute of Neurological Disorders and Stroke. It was also supported by Children Living with Inherited Metabolic Disease and the Juanma Fund.
Tags: animal research, biosciences, brain, health, research, Waisman Center
-
Interwoven
How Indigenous knowledge and science can work together to communicate about climate.
-
UW–Madison’s reach throughout Wisconsin adds up to $38.9 billion a year
The university supports more than 287,000 jobs and helps drive private-sector growth.
-
‘Being part of something bigger than you’ with Badger Volunteers
UW–Madison students head into the local community to find purpose and make a difference.
-
UW–Madison’s Tech Exploration Lab: Where the classroom meets the real world
The lab is built around a simple expectation: Students come to build, test and refine projects with real problems in mind.



