
Deleting a gene prevents Type 1 diabetes in mice by disguising insulin-producing cells
Removing a gene from the cells that produce insulin prevents mice from developing Type 1 diabetes by sparing the cells an attack from their own immune system, a new UW–Madison study shows.
The cellular sleight of hand may suggest ways to prevent Type 1 diabetes in high-risk individuals, as well as other diseases in which the immune system targets the body’s own cells.

Feyza Engin
People with Type 1 diabetes — once called juvenile diabetes — make little or no insulin, a hormone necessary to make energy from the sugar in their blood. At an early stage in the disease, their immune system’s frontline soldiers, called T cells, incorrectly identify insulin-producing beta cells as a threat and kill them, leading to complete insulin deficiency.
The resulting chaos must be managed for the rest of a patient’s life with diet, blood sugar measurement and insulin shots. Type 1 diabetes afflicts as many as 20 million people around the world, contributing to glaucoma, nerve damage, high blood pressure and stroke. In the U.S., it shortens life expectancy by more than a decade.
“The thing is, individuals who are at high risk can be identified,” says Feyza Engin, a biomolecular chemistry professor at the University of Wisconsin–Madison and lead author of a new study published today in the journal Cell Metabolism. “They have autoantibodies in their blood serum, meaning we can actually tell who is going to develop Type 1 diabetes within a couple of years. But there’s not much for clinicians to do but send them home, because there’s no cure for Type 1 diabetes.”
Engin’s lab altered a line of mice genetically destined to develop Type 1 diabetes. Right before the immune attack usually begins, they removed from the beta cells alone a gene called IRE1-alpha, involved in the mouse cells’ response to stress.
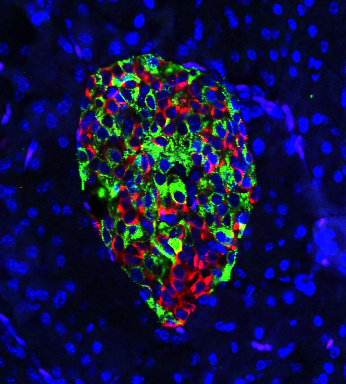
Cells in the pancreas of a gene-edited mouse produce nearly equal amounts of the hormones insulin (green) and glucagon (red). In mice that develop Type 1 diabetes, green would predominate, and insulin production would draw the deadly attention of a disordered immune system. Courtesy of Hugo Lee
Engin expected removing this gene in insulin-producing cells would lead to accelerated diabetes. But the gene removal made a striking and unexpected difference in the mice.
“We expected the beta cells would die soon,” Engin said. “Instead, my students told me that the blood glucose levels of the mice were becoming normal following an initial increase lasting a couple of weeks. I couldn’t believe it. I said, ‘What? No. Just measure it one more time.’”
The beta cells were indeed becoming normal insulin producers. But first they were taking a step backward into immaturity.
“Once we remove this gene, it’s almost like the beta cells are undergoing a disguise,” says Engin, who was joined by first author Hugo Lee, a graduate student, in publishing the results. “They lose their mature identity. They de-differentiate and exhibit features of progenitor cells, and express hormones of other cell types in addition to insulin.”
If that de-differentiation happens before an auto-immune response puts the beta cells in danger, the T cells they meet respond differently.
The Engin lab’s new findings could help guide the way candidate diabetes drugs in clinical trials are used, or help create new therapies. And they may have a similar effect in other auto-immune disorders — like arthritis, lupus and multiple sclerosis.
“When they de-differentiate, they don’t act like typical beta cells anymore. They reduce the expression of many genes that signal to immune cells, ‘Come and eat me!’” Engin says. “Those signals go down, and that actually is altering the diabetogenic activity of T cells. They don’t really recognize the beta cells as a problem anymore. They don’t attack.”
And then, just as importantly, the immature, de-differentiated beta cells re-differentiate into functional, mature beta cells.

Hugo Lee
“The mice experienced a little transient hyperglycemia. They have relatively high blood sugar, which isn’t dangerous, for a few weeks,” says Engin, whose lab is supported by the National Institutes of Health and the Juvenile Diabetes Research Foundation. “But then the beta cells get back to work, and make insulin like they’re supposed to.”
The T cells alter their activity and stick with the change, leaving the beta cells alone for as long as the lab has followed the mice so far.
“That’s the beauty of it,” Engin says. “Even after the beta cells come back, the T cells leave them alone. They still have no diabetogenic activity one year later, which is like 40 or 50 years in a human life.”
Two drugs being tested in clinical trials for Type 1 diabetes target the stress response of beta cells — including a drug whose efficacy Engin discovered in mice while working at Harvard University. Her lab’s new findings could help guide the way candidate diabetes drugs in clinical trials are used, or help create new therapies. And they may have a similar effect in other auto-immune disorders — like arthritis, lupus and multiple sclerosis — in which a particular cell type’s activity draws dysfunctional immune attention.
“We’ve found a very important time point where de-differentiation helps greatly reduce the immune cells’ diabetogenic activity,” Engin says. “If you can determine an appropriate cell targeted by auto-immune response, and make those victim cells less functional, less mature in the beginning, maybe they can avoid their role in the progress of other diseases, too.”
Tags: animal research, chemistry, diabetes, health
-
Interwoven
How Indigenous knowledge and science can work together to communicate about climate.
-
UW–Madison’s reach throughout Wisconsin adds up to $38.9 billion a year
The university supports more than 287,000 jobs and helps drive private-sector growth.
-
‘Being part of something bigger than you’ with Badger Volunteers
UW–Madison students head into the local community to find purpose and make a difference.
-
UW–Madison’s Tech Exploration Lab: Where the classroom meets the real world
The lab is built around a simple expectation: Students come to build, test and refine projects with real problems in mind.



