
Discovery reveals mitochondria as potential treatment target for fragile X syndrome
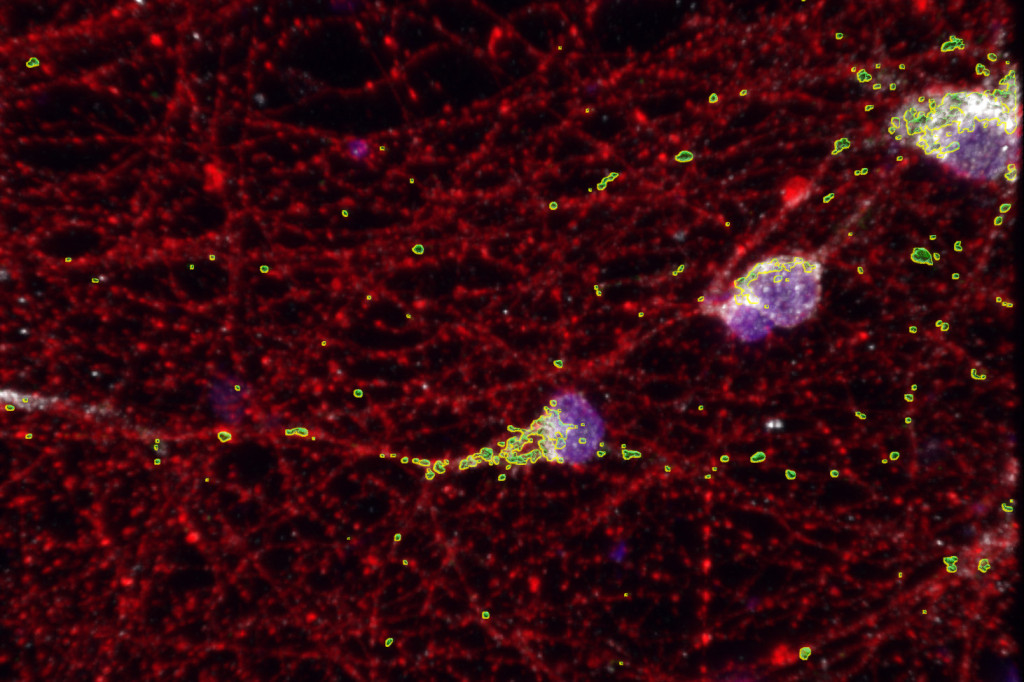
The energy-making organelles called mitochondria (shown in green) that work inside cells to make energy aren’t working as they should in the neurons (shown in red) of people with fragile X syndrome. UW–Madison researchers have identified a protein and gene involved in this mitochondrial dysfunction, as well as a potential treatment. Image by: Minjie Shen
Fragile X syndrome, the most common form of inherited intellectual disability, may be unfolding in brain cells even before birth, despite typically going undiagnosed until age 3 or later.
A new study published today in the journal Neuron by researchers at the University of Wisconsin–Madison showed that FMRP, a protein deficient in individuals with fragile X syndrome, has a role in the function of mitochondria, part of a cell that produces energy, during prenatal development. Their results fundamentally change how scientists understand the developmental origins of fragile X syndrome and suggest a potential treatment for brain cells damaged by the dysfunction.

Xinyu Zhao is a neuroscience professor and neurodevelopmental diseases researcher at UW–Madison’s Waisman Center. Four postdoctoral fellows in her lab led the study.
The study, led by four postdoctoral fellows — Minjie Shen, Carissa Sirois, Yu (Kristy) Guo and Meng Li — working in the lab of the lab of Xinyu Zhao, neuroscience professor and neurodevelopmental diseases researcher at UW–Madison’s Waisman Center, found FMRP regulating a gene called RACK1 to promote mitochondrial function. Using a drug to enhance mitochondrial function, they were able to rescue brain cells damaged by lack of FMRP.
Individuals with FXS may present developmental delays — not sitting, walking or talking at expected ages — as well as mild to severe intellectual disability, learning disabilities and social and behavioral problems. About half are also diagnosed with autism spectrum disorder.
In previous research, Zhao found that mitochondria in mice with an FMRP deficiency that imitates FXS were smaller and unhealthy. Diving deeper, they also discovered that FMRP regulates genes involved in mitochondria fission-fusion, a process in which mitochondria fuse into a bigger shape in order to produce more energy for the cell.
For the study, researchers grew brain cells called neurons grown from induced pluripotent stem cells. Because the stem cells came from people with FXS, the researchers could study the development of the disorder at a cellular level, determining whether mitochondria in human cells experienced issues similar to those in mice.
“And indeed, we found that human neurons also have fragmented (smaller) mitochondria,” Zhao says. They also found fewer mitochondria in neurons derived from FXS patients, which they did not see in the neurons of the mice modeling FXS.
“In human neurons, it’s a deficit in twofold. Not just fission-fusion, but also likely in the production of mitochondria,” Zhao says.
Although it has long been known that FMRP is deeply involved in FXS, the new discovery pinpoints a role for the protein in early development of the condition.
Symptoms of FXS present long after the baby is born. Many babies appear to be developing typically before showing slower development, autistic features or developmental deficits. Children with FXS are typically diagnosed at three years of age or older.
“Which means many scientists have been thinking that FMRP is more important for the postnatal maturation state,” Zhao says.
FMRP is protein that regulates the use of messenger RNA, sort of a working copy of DNA used to produce the proteins that make things happen in cells. The researchers found that many of the mRNA strands that interact with FMRP are implicated in autism, providing a molecular link between FXS and autism spectrum disorder. Unexpectedly, many FMRP-bound mRNAs are expressed by genes classified as essential — genes that are very busy during prenatal development but less active after birth.
“This means that FMRP has a function in prenatal development that we have not really thought about before,” Zhao says. “The fact that we found that FMRP also regulates prenatal development is really interesting and is actually indicating that what we see in fragile X syndrome, some of the effects already happened within the prenatal development.”
One of those essential genes is RACK1, identified for the first time as playing a role in FXS.
“When RACK1 is lower in fragile X neurons, the mitochondria are suffering and the neurons exhibit mitochondrial deficit and hyperexcitability, like immature neurons. But when we reintroduce RACK1, we can rescue this,” Zhao says.
Using cultured neurons derived from individuals with FXS to screen for drugs, the researchers found a drug called leflunomide that corrected mitochondrial deficits. The treatment improved mitochondrial function and reduced the neurons’ hyperexcitability.
Next, Zhao wants to do a detailed biochemical analysis of mitochondrial dysfunction and figure out which key proteins are less present in FXS-affected neurons. She is also working on better understanding how RACK1 and leflunomide work to rescue mitochondrial function.
Other collaborators on the study include Waisman Center investigators Qiang Chang, Anita Bhattacharyya, Andre Sousa, Daifeng Wang, Donna Werling and UW–Madison neuroscience professor Jon Levine.
This research was supported by grants from the National Institutes of Health (R01MH118827, R01NS105200, R01MH116582, R01MH118827, R01HD064743, R01NS064025, R01AG067025, U01MH116492, P51 OD011106, U54HD090256, P50HD105353, R24HD000836 and T32 GM141013) and the Department of Defense (W81XWH-22-1-0621), as well as funding from the Brain Research Foundation, Wisconsin Alumni Research Foundation, Brain and Behavior Research Foundation, Simons Foundation, FRAXA Research Foundation, Autism Science Foundation and UW–Madison awards including the Jenni and Kyle Professorship, Vilas Faculty Mid-Career Investigator Award, Kellett Mid-Career Award, SciMed scholarships, Stem Cell and Regenerative Medicine Center postdoctoral fellowship and Hilldale Undergraduate Research Fellowship.
Tags: nervous system, research, stem cells, Waisman Center
-
Interwoven
How Indigenous knowledge and science can work together to communicate about climate.
-
UW–Madison’s reach throughout Wisconsin adds up to $38.9 billion a year
The university supports more than 287,000 jobs and helps drive private-sector growth.
-
‘Being part of something bigger than you’ with Badger Volunteers
UW–Madison students head into the local community to find purpose and make a difference.
-
UW–Madison’s Tech Exploration Lab: Where the classroom meets the real world
The lab is built around a simple expectation: Students come to build, test and refine projects with real problems in mind.



